Deepen your understanding of the genetic dynamics of urea
cycle disorders (UCDs).


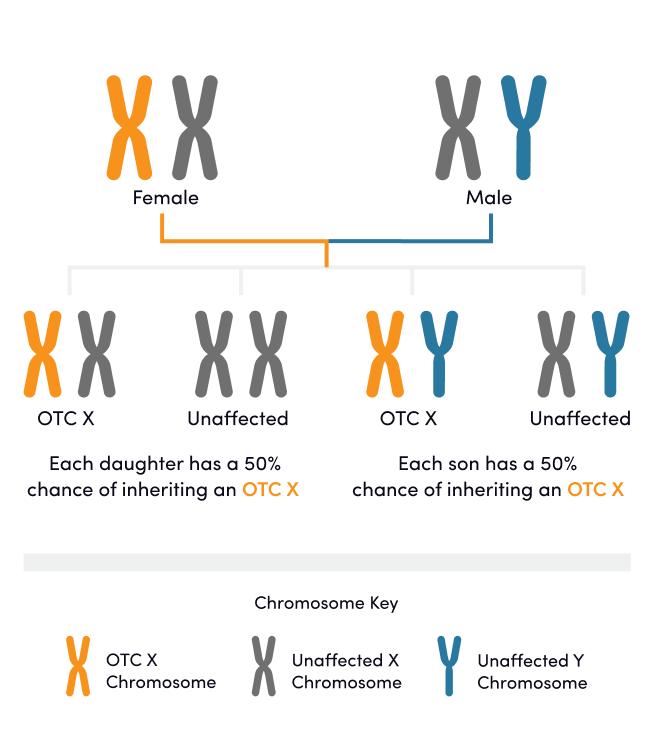
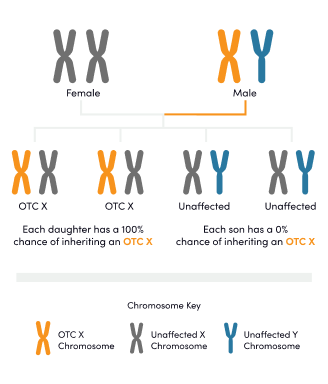
Ornithine transcarbamylase (OTC) deficiency is the only X-linked UCD subtype. A mother may pass the affected OTC gene to her sons or daughters, while a father may pass the affected OTC gene to his daughter.1 Individuals with OTC deficiency may experience symptoms and may require treatment.1-3
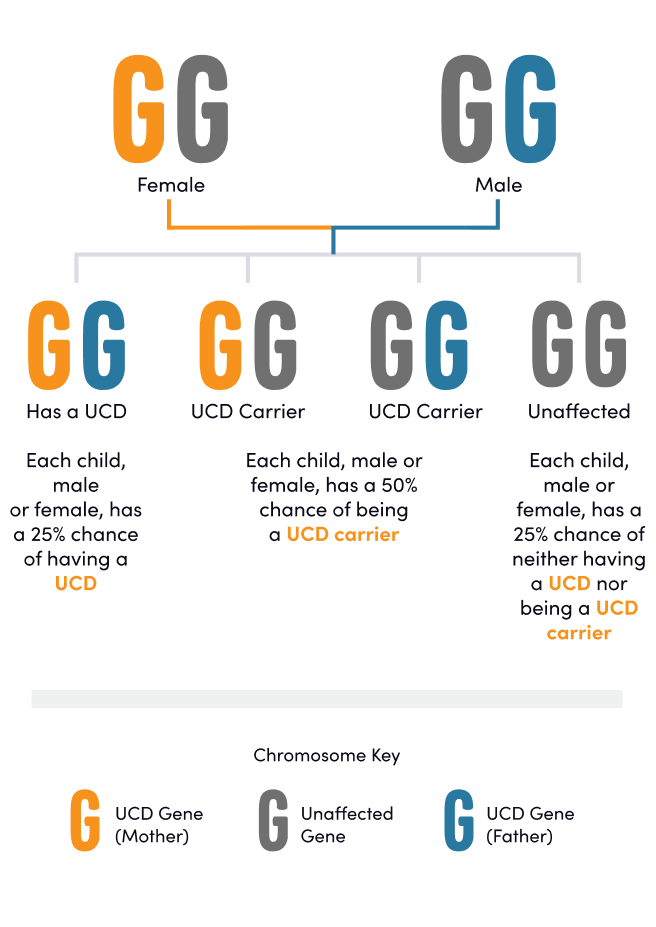
All other UCD subtypes are inherited in an autosomal recessive manner.1 Individuals with these UCD subtypes have a lower likelihood of experiencing symptoms; however, the presentation and severity of symptoms in individuals with an autosomal recessive UCD may vary based on the specific UCD subtype.1
Rarely, a UCD is the result of a new or de novo mutation.1 These mutations, similar to inherited mutations, may be passed on to children.1



Identifying symptoms can help expedite a diagnosis
Resources are available to help your patients understand their diagnosis
1. Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea cycle disorders overview. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; April 29, 2003. 2. Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42(6):1192-1230. 3. National Organization for Rare Disorders (NORD). The Physician’s Guide to Urea Cycle Disorders. Accessed March 23, 2020. https://rarediseases.org/physician-guide/urea-cycle-disorders